
1. O que é a Síndrome de Deleção 22q11.2?
É uma síndrome causada por falta de uma parte da informação genética no cromossomo 22, em uma pessoa.
Outros nomes comuns da mesma síndrome são:
- Síndrome de DiGeorge
- Síndrome Velocardiofacial
2. O que é informação genética?
O que torna uma pessoa diferente da outra é a informação genética (DNA) que ela carrega dentro de suas células. Toda a informação genética de uma pessoa (todo o seu DNA) está organizada na forma de cromossomos.
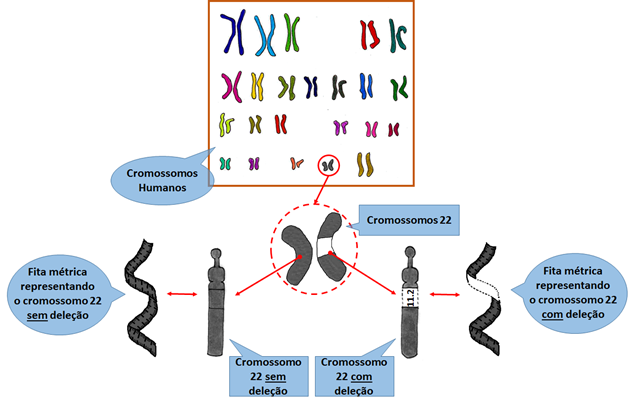
Para compreender melhor, imagine que essa informação está organizada como se fossem 23 pares (= 46) de fitas métricas. Cada “fita métrica” representa um cromossomo e, nos centímetros da “fita” estariam presentes as informações necessárias para o desenvolvimento, crescimento e funcionamento do corpo por toda a vida de uma pessoa.
Em cada par, uma “fita métrica” vem da mãe e a outra do pai. Então, para o funcionamento perfeito da informação genética, precisamos ter as duas cópias da “fita”, da mãe e do pai.
Na Síndrome de deleção 22q11.2 parte de um dos cromossomos 22 se perde, ou seja, é como se uma das “fitas métricas” estivesse com alguns centímetros faltando. Assim, certas informações importantes que deveriam estar com duas cópias (pois as fitas estão em pares) estão com apenas uma cópia, resultando nos sinais e sintomas dessa síndrome (ver figura 1).
Figura 1 - Ilustração representando os 23 pares de cromossomos.
3. A síndrome é comum?
É uma síndrome rara. Calcula-se que exista uma pessoa com essa síndrome entre cada 2.000 a 4.000 pessoas. Porém, entre as diferentes síndromes de deleção (pois existem outras) a Síndrome de deleção 22q11.2 é a mais comum no mundo.
4. Quando suspeitar?
Existem algumas “pistas” para que o médico suspeite que uma pessoa tem a Síndrome de deleção 22q11.2. Elas são as seguintes:
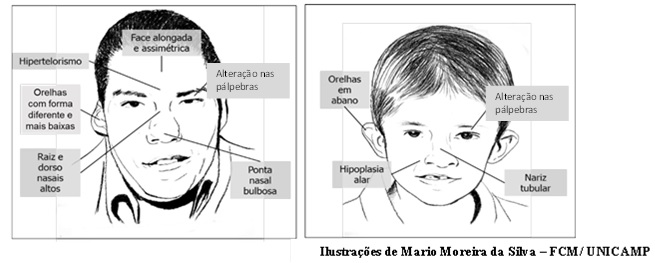
- Sinais na cabeça e no rosto: rosto alongado, raiz nasal (parte do nariz entre os olhos) mais alta e ponta do nariz bulbosa (mais arredondada), alteração nas pálpebras, orelhas com forma diferente, olhos mais afastados um do outro, boca pequena e queixo pequeno.
- Figura 2 (Ilustrações de Mario Moreira da Silva – FCM/ UNICAMP):
- Ilustração baseada em fotos de pessoas com a Síndrome de deleção 22q11.2.
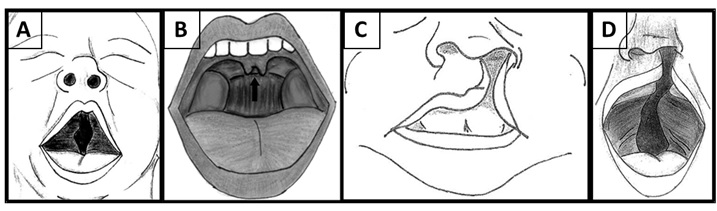
- Defeitos no céu da boca: insuficiência velofaríngea, que causa voz fanhosa e dificuldade para engolir; fenda palatina – céu da boca “aberto” (figura 3 - A), úvula (campainha no céu da boca) bífida (dividida ao meio) (figura 3 - B). Algumas vezes, podem também acontecer fenda labial (figura 3 - C) ou fenda labial e palatina (figura 3 - D). Esses defeitos podem dificultar a alimentação da criança, que pode apresentar refluxo, queimação ou vômito após as refeições.
Figura 3: Ilustração dos defeitos que podem acontecer na boca.
- Problemas no coração: o tipo de defeito pode variar desde um sopro no coração sem consequências sérias até defeitos graves que precisam de cirurgia.
- Falta de cálcio: pode causar convulsões (ataque epiléptico) em bebês e crianças e movimentos musculares involuntários (que acontecem sem querer) em adultos.
- Deficiência no sistema imunológico: a criança fica doente “toda hora” (problema no sistema de defesa do organismo), principalmente com gripes e outras doenças respiratórias;
- Atraso no desenvolvimento: a criança demora mais para sentar, andar, falar, aprender na escola.
Outros sinais menos comuns também podem estar presentes, como: perda auditiva (escuta mal), escoliose ou outros problemas nos ossos, problemas hormonais (ex: hipertireoidismo, hipotireoidismo), alterações da formação dos olhos, entre outros.
É importante saber que apesar dessas características citadas ocorrerem em pessoas com a síndrome, nem todos apresentarão todas essas características juntas. Apenas a presença de uma ou poucas delas pode ser um motivo para o médico suspeitar dessa síndrome.
5. Como confirmar o diagnóstico?
Para ter certeza que uma pessoa tem essa síndrome é preciso realizar alguns exames de laboratório. Isso é necessário porque os sinais e sintomas da síndrome são bem variados, o que dificulta o diagnóstico clínico (apenas com a avaliação do médico). Por isso, o médico deve pedir o teste de laboratório, que é feito com sangue. Nele, observa-se a quantidade de material do cromossomo 22, como se fosse observar os centímetros das fitas métricas, se essas estão completas ou com alguns centímetros faltando.
6. De onde vem a síndrome de deleção 22q11.2?
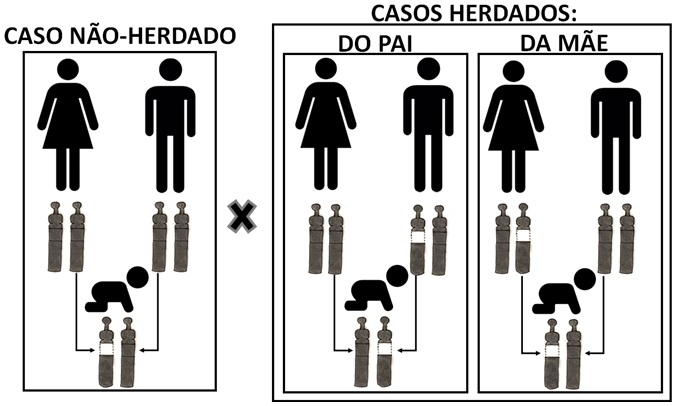
Na maioria dos casos (85%), acontece por acaso, não vem nem do pai e nem da mãe (ver figura 4);
Em 15% dos casos o pai ou a mãe pode ter a mesma alteração (ver figura 4);
Observação: Quando confirmado o diagnóstico da pessoa, a investigação dos pais deve ser realizada para poder dizer se existe risco para outros filhos. Com o exame em mãos, os pais precisam de uma consulta de aconselhamento genético. Este precisa ser feito por profissional preparado para isso, pois cada situação pode ter diferentes possibilidades.
Figura 4: Ilustração diferenciando caso não herdado de herdado.
7. Se uma pessoa apresenta a síndrome com sintomas leves, singnifica dizer que outras pessoas desta família que apresentarem a síndrome também terão sintomas leves?
Não. Existe uma variação muito grande na apresentação dos sintomas, mesmo entre membros da mesma família.
8. Por que aconteceu?
Nos casos em que a deleção no cromossomo 22 não veio dos pais, aconteceu por acaso. Ou seja, não tem relação com fatores do ambiente (como nervoso, remédio que a mãe tomou, comida ou outra vontade que a mãe passou na gravidez, por exemplo).
9. Qual a chance de acontecer novamente?
Se a deleção aconteceu por acaso (pais sem a deleção): O risco de acontecer novamente em irmãos da pessoa com a deleção é menor do que 1%.
Risco para quem tem a deleção: O risco da pessoa que tem a deleção transmitir para um filho é de 50% para cada filho que a pessoa tiver.
Observação: Este risco precisa ser avaliado em cada família em uma consulta de aconselhamento genético. Nela, além do risco, o especialista vai verificar outros fatores genéticos importantes e realizar uma orientação específica para os pais e irmãos.
10. Como deve ser o tratamento e quais os acompanhamentos médicos necessários?
Com o diagnóstico confirmado, a pessoa deverá receber tratamento especializado para ter uma qualidade de vida melhor.
O tratamento deve ser:
- INDIVIDUALIZADO, ou seja, varia de acordo com as necessidades e problemas de saúde que cada pessoa apresenta.
- MULTIDISCIPLINAR, ou seja, necessita da participação de diversos profissionais para acompanhar a pessoa com a síndrome.
Os principais profissionais que podem ser necessários para o tratamento são: Pediatra, Geneticista, Cardiologista, Endocrinologista, Imunologista, Oftalmologista, Otorrinolaringologista, Fonoaudiólogo, Ortopedista, Dentista, Psicólogo, Psiquiatra, Psicopedagogo, entre outros.
11. O que pode acontecer com quem tem a síndrome?
Observação: Essas são as características mais comuns das pessoas com esta síndrome. Porém, isso não significa que todos apresentam essa evolução.
A pessoa que nasce com a síndrome pode ter problemas de saúde ao longo da vida, mas eles não estão todos visíveis ao nascimento. Alguns sinais e sintomas dependem da idade da pessoa para aparecer. Por exemplo, o problema de coração pode estar presente ao nascimento, mas a dificuldade de aprender só aparece na idade escolar.
Figura 5: Principais problemas apresentados de acordo com a idade:
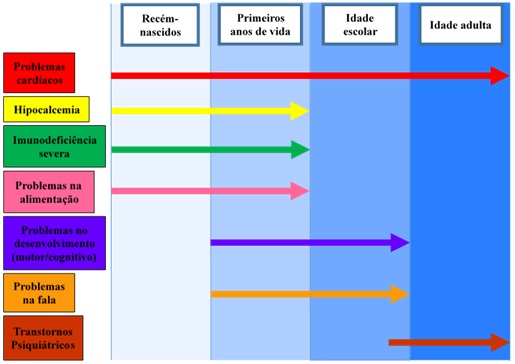
Geralmente os portadores da síndrome apresentam estas características:
- - Demoram mais para firmar o pescoço e a cabeça, sentar, engatinhar, andar e realizar outras habilidades motoras;
- - Demoram mais para falar e, quando falam, podem ter a voz fanhosa (anasalada);
- - Tem altura menor que as demais crianças da mesma idade;
- - Apresentam dificuldades para aprender na escola;
- - Apresentam dificuldade para se concentrar e para manter a atenção;
O tratamento com estimulação global precoce ajuda muito o desenvolvimento do bebê, assim como os profissionais que trabalham no apoio para aprendizagem (psicopedagogia e terapeuta ocupacional, por exemplo).
Apesar das dificuldades, muitas pessoas com essa síndrome conseguem viver bem e se inserir no mercado de trabalho.
Você pode convidar o médico que cuida da pessoa com a síndrome, a visitar o site do Projeto Crânio-Face Brasil e ler o “Guia de Manejo Clínico para Pacientes com a Síndrome da Deleção 22q11.2”, disponível na área de material destinado a profissionais de saúde.
Você também pode baixar este material em pdf: http://www.fcm.unicamp.br/fcm/sites/default/files/2016/page/guia_-_pacie...